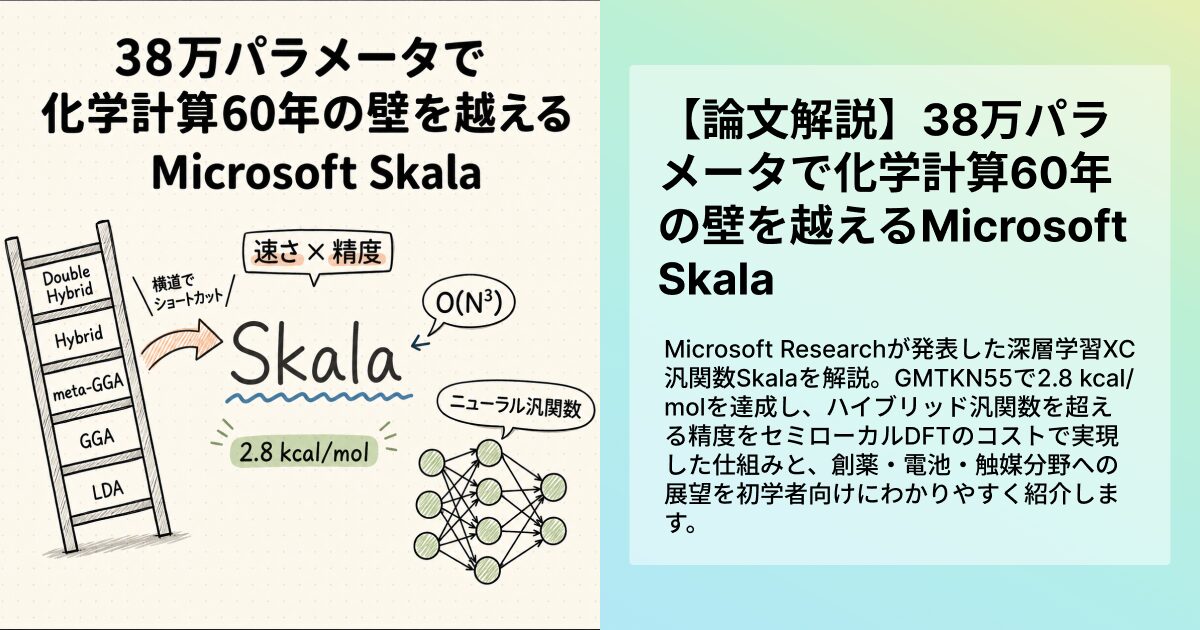
AIが分子の世界に踏み込んだ日 Skalaって何がすごいの?
2026年4月、ある論文がひっそりと更新されました。タイトルは「Accurate and scalable exchange-correlation with deep learning」。日本語にすると「深層学習による高精度かつスケーラブルな交換相関」となります。Microsoft Researchが公開した、わずか38万パラメータの小さなニューラルネットワークが、化学計算の世界で60年続いてきた「速さと精度のどちらかしか取れない」というジレンマを破ったという話です。
この小さなニューラルネットには、Skalaという名前がついています。
「化学計算?私には関係ないかも」と思った方、少しだけ待ってください。スマホのバッテリーを長持ちさせる新しい電池材料、副作用の少ない新薬、二酸化炭素を効率よく吸収する触媒。これらの研究開発の舞台裏では、毎日のようにコンピュータが分子の中の電子のふるまいを計算しています。その計算を支えてきた手法が「DFT(密度汎関数理論)」と呼ばれるもので、Skalaはその核となる部分をAIで置き換える試みです。
私はこの論文を読みながら、ちょっと不思議な気持ちになりました。なぜなら、こんな比喩が浮かんできたからです。
DFTという60年使われてきた計算手法を「階段」にたとえると、Skalaは階段を上ったわけではなく、階段の横にトンネルを掘ったような存在なんじゃないか。
本記事では、論文の中身を中学生にもイメージできる言葉でほぐしながら、Skalaが計算化学にもたらした衝撃と、これから私たちの暮らしに広がりそうな影響について、じっくり追いかけていきます。難しそうに見える内容ですが、最後まで読み終えるころには「ああ、つまりこういうことか」と腑に落ちる構成にしました。
そもそもDFTって何をするものなの?
DFTは「Density Functional Theory」の頭文字で、日本語では「密度汎関数理論」と呼びます。物質の性質を、コンピュータ上で量子力学にもとづいて予測する計算手法のひとつです。
私たちの身の回りにあるすべての物質は、原子核と電子からできています。電子のふるまいが分子の性質や化学反応を決めるので、新しい薬や電池材料を設計するには、電子のふるまいを正確に知る必要があります。けれど電子は量子力学にしたがって動くため、ひとつひとつ追いかけようとすると計算量が爆発してしまうのです。
Matlantisの解説記事では、DFTの考え方をこう説明しています。「個々の電子の波動関数を全部追う代わりに、空間内のどこにどれくらい電子が集まっているか(電子密度)からエネルギーを計算しよう」というアイデアです。これが1960年代に生まれて、1998年にはノーベル化学賞を受賞しました。今では世界中の研究者が日常的に使っている、量子化学の中核ツールになっています。
Skalaの「ここがすごい」を一行で言うと
論文のarXiv要約に書かれているフレーズをそのまま借りるとこうなります。「ハイブリッド汎関数を超える精度を、セミローカルDFTのコストで実現した」。
専門用語が並んでいて怖く見えますが、噛み砕くとこういう話です。
- 「ハイブリッド汎関数」は、これまで化学計算で「精度は高いけど計算量が10倍から100倍重い」とされてきた方法
- 「セミローカルDFT」は、「精度は中くらいだけど高速」とされてきた方法
この2つは長らく「精度を取るか、速さを取るか」の二者択一でした。Skalaはその二者択一そのものを成り立たなくしたのです。具体的には、化学計算のオリンピックとも呼ばれるGMTKN55というベンチマークで、平均誤差2.80 kcal/molを達成しました(次のセクション以降で詳しく説明します)。
「速さは中くらいだけど精度は最高クラス」という、これまで存在しなかった選択肢が登場した、ということです。
そしてもうひとつ、注目すべきポイントがあります。Skalaのパラメータ数はわずか385,217個。最近話題のChatGPTやGeminiなどの大規模言語モデルが数千億パラメータを持つことを思えば、まさに「小さな巨人」です。なぜそんな小さなモデルでこんな成果が出せたのか。次のセクションから、その秘密に少しずつ近づいていきましょう。
ヤコブの梯子と60年続いた「精度と速さの呪い」
前のセクションで、DFTは「電子密度からエネルギーを計算する」手法だとお伝えしました。けれど、実はDFTには60年来の頭の痛い問題が残されてきました。それを理解するために、まずDFTの計算式の中で「どの部分」が問題なのかを掘り下げてみましょう。
交換相関エネルギー、それは「式の形がわからない部分」
DFTの計算式は、ざっくり次のような構成になっています。
- 電子と原子核のクーロン相互作用(電気的な引力や斥力):式の形は完全にわかる
- 電子の運動エネルギー:これも近似的にわかる
- 電子同士のハートリー相互作用(電子雲が作る電場):これもわかる
- そして「交換相関エネルギー」:式の形が、厳密にはわかっていない
最後の「交換相関エネルギー」は、電子同士の量子力学的な「絡み合い」を表す部分です。電子は同じ場所に2個入れない(パウリの排他律)とか、お互いに距離をとりたがる(クーロン斥力)とか、そんな複雑な振る舞いを反映しています。
この部分を「交換相関汎関数」という関数で近似するのですが、その「正解の関数」は数学的にはまだ誰も知りません。だから、計算化学者たちは長年、この近似式をどう設計するかに知恵を絞ってきました。
ヤコブの梯子という名づけのうまさ
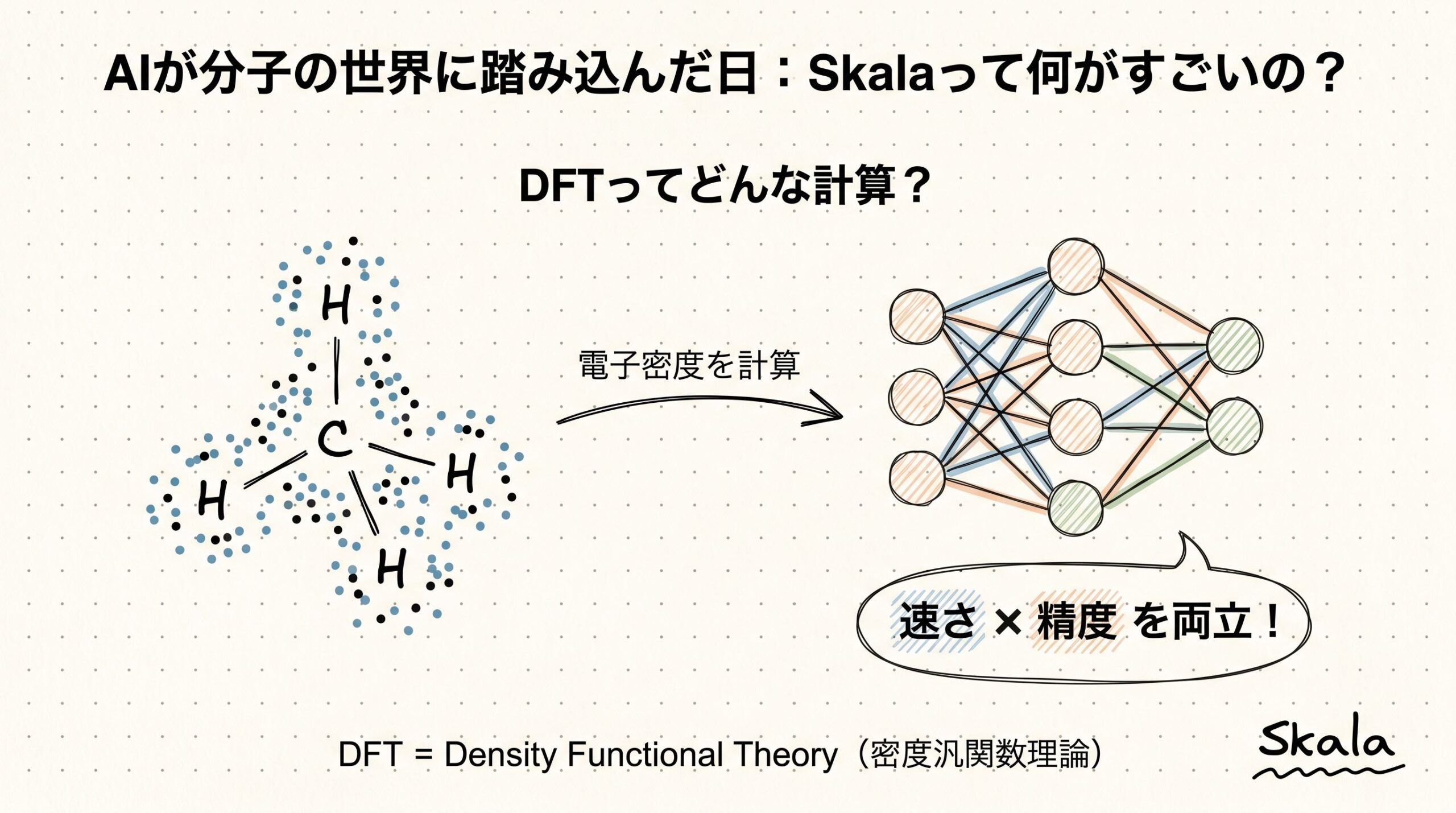
2001年、物理学者のJohn Perdewは、近似汎関数の階層構造を「ヤコブの梯子(Jacob’s Ladder)」と名づけました。旧約聖書に出てくる、地上から天に届く梯子のイメージです。地上は「化学的精度の天国」とは程遠い世界、天は「実験と完全に一致する計算精度」の世界。研究者たちは梯子を一段ずつ上りながら、天国に近づこうとしてきました。
梯子は5段あります。下から順に紹介します。
1段目は LDA(局所密度近似)。電子密度の値だけを使う、もっともシンプルな近似です。1段目だけあって、精度はそこそこですが速い。
2段目は GGA(一般化勾配近似)。電子密度の「変化の急さ」(密度勾配)も使うようにした近似です。代表的なのはPBE汎関数で、材料科学の業界では事実上の標準になっています。
3段目は meta-GGA。さらに「運動エネルギー密度」という情報も加えました。SCAN汎関数などが代表例です。
4段目は Hybrid汎関数。ここで雰囲気が変わります。Hartree-Fock法という別の量子化学手法から「厳密交換項」と呼ばれる正確な部分を一定割合で混ぜるのです。B3LYPやPBE0、ωB97M-Vなどがここに入ります。精度は跳ね上がるけれど、計算量も跳ね上がります。
5段目は Double Hybrid汎関数。MP2(メラー・プレセット2次摂動論)の相関項まで取り込みます。最高精度クラスですが、計算量はさらに重くなります。
「梯子を上ると、計算時間も天国に届く」皮肉
ここに60年来の呪いがあります。梯子を上るほど精度は上がるのですが、それと引き換えに計算時間が爆発的に増えてしまうのです。
DFTの計算時間は、電子数Nに対しておおよそO(N³)、つまり原子数を2倍にすると計算時間が8倍になる関係です(論文本文より)。これがセミローカルなLDA、GGA、meta-GGAの世界。けれど、4段目のHybridに進むと厳密交換項の計算が必要になり、O(N⁴)を超えるオーダーになります。原子数が増えれば10倍から100倍も時間がかかるのです。
5段目のDouble Hybridともなれば、O(N⁵)以上。100原子を超える分子では、1回の計算に数日から数週間かかることも珍しくありません。これが「実験並みの信頼性が欲しいなら、計算時間も実験並み(数日待ち)」という皮肉な状況を生んできたのです。
「化学的精度1 kcal/mol」という遠い目標
計算化学の世界には「化学的精度(chemical accuracy)」という言葉があります。実験と理論計算の誤差が約1 kcal/mol未満という基準です。
なぜ1 kcal/molなのか。室温で平衡状態にある化学反応を考えると、わずか1.3 kcal/molのエネルギー差で、生成物と反応物の存在比が10倍も変わるからです(Matlantisの解説より)。つまり、実験を予測したいなら、最低でもこのレベルの精度が必要です。
ところが、これまでの汎関数で化学的精度を出すには、5段目のDouble Hybridを使うしかありませんでした。「精度は欲しいけど、計算時間が現実的じゃない」というジレンマが、新薬や新材料の開発スピードを阻んできた最大の壁です。
2017年に発表されたGMTKN55ベンチマークでは、約220個の汎関数が評価されました。最高峰のDouble HybridであるDSD-PBEP86でも誤差は約2 kcal/mol、Hybrid級のωB97M-Vで3.23 kcal/molという結果でした。
「梯子を上っても、まだ1 kcal/molには届かない」。これが2025年までの計算化学の現状でした。
では、Skalaは梯子の何段目に位置するのでしょうか。実は、Skalaは梯子の3段目(meta-GGA)の入力情報しか使っていません。それなのに、4段目のHybridを超える精度を出してしまった。これがどれほど驚くべきことか、次のセクションでアーキテクチャの中身に踏み込みながら見ていきましょう。
Skalaの仕組み 格子点同士でおしゃべりする小さなニューラルネット
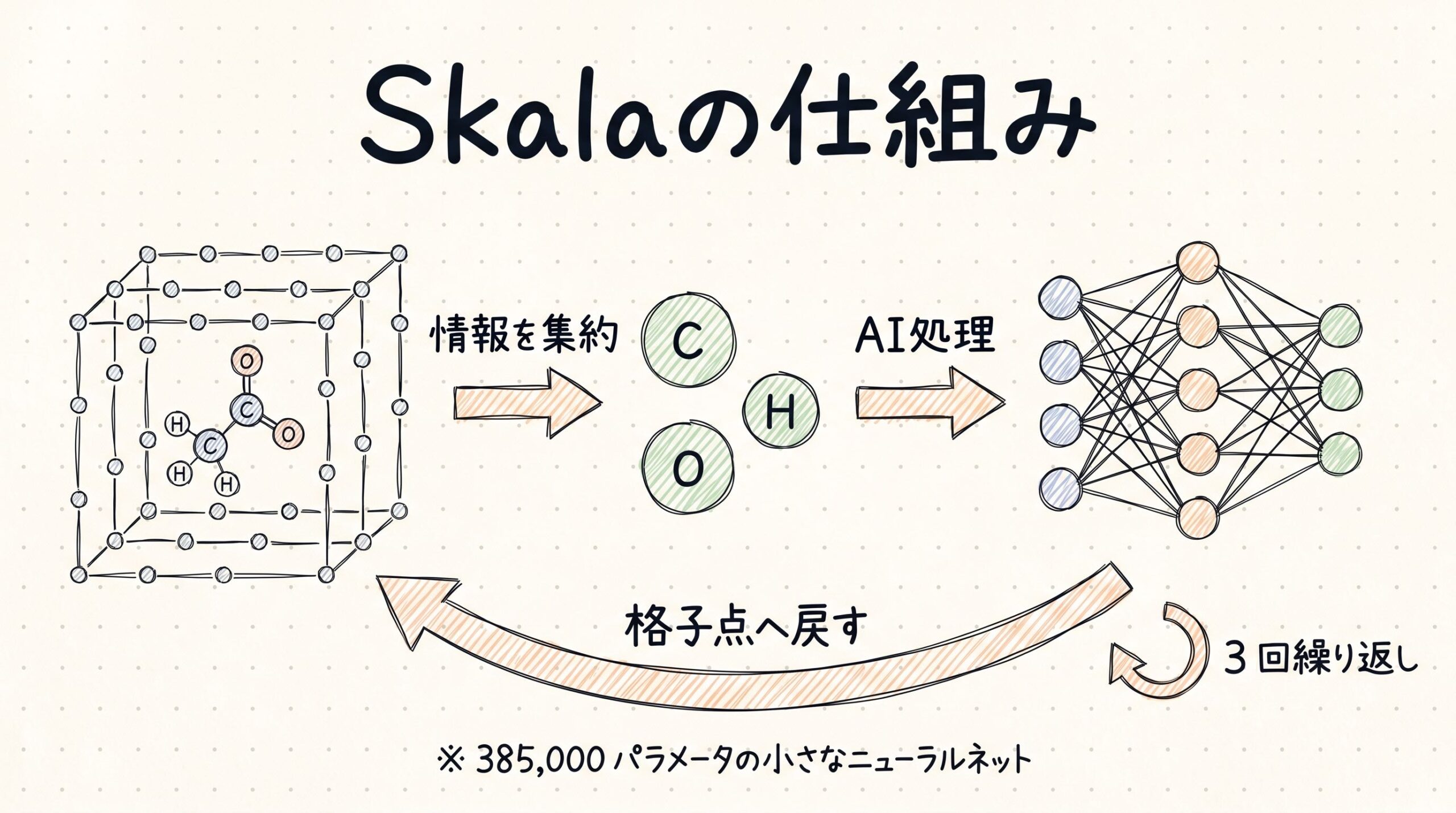
前のセクションで、ヤコブの梯子の3段目までは「速いけど精度がそこそこ」、4段目以降は「精度が高いけど重い」というトレードオフが60年続いてきたことをお伝えしました。Skalaはこの梯子を上らずに、横にショートカットを掘った存在です。では、どうやってそれを実現したのでしょうか。
「手で式を作る」から「データから式を学ぶ」への転換
従来の汎関数は、すべて研究者が手で式を設計してきました。「電子密度のこの部分はこんな形のべき関数で近似しよう」「ここはガウス関数を使おう」といった具合に、物理的・化学的な直感や、特定の分子で実験値とのフィットが良くなるように、職人芸で組み立てられてきました。
Skalaはこの発想を捨てています。論文の要約にはこう書かれています。「expensive hand-designed features を bypass し、データから直接、表現を学習する」。
簡単に言えば、こういう違いです。
- 従来:人間が「こんな数式が良さそうだ」と仮説を立てて式を作る
- Skala:高精度な参照データを大量に集め、ニューラルネットに「この入力からこの出力を出せるように学習しなさい」と任せる
この発想の転換は、画像認識の世界で起きた変化とそっくりです。かつての画像認識では、SIFTやHOGといった「人間が設計した特徴量」が主流でした。それがCNN(畳み込みニューラルネットワーク)の登場で、生のピクセルから直接学習する方式に置き換わりました。Skalaは、計算化学にとっての「CNN登場の瞬間」と言えるかもしれません。
たった38万パラメータの理由
少し驚くのは、Skalaのパラメータ数がわずか385,217個である点です。最近のChatGPTやGeminiが数千億パラメータを持つことを思えば、ほとんど誤差のような数字です。
なぜこんなに小さくて済むのか。理由は、Skalaが「最初から物理に詳しい」状態でスタートしているからです。
Skalaの入力は、meta-GGAレベルの情報、つまり「電子密度」「電子密度の勾配」「Kohn-Sham運動エネルギー密度」の3種類です(Hugging Faceのモデルカードより)。これらは何十年もの計算化学の知見が詰まった、すでに「物理を知っている特徴量」です。Skalaは、その既知の特徴量を「どう組み合わせれば本当のエネルギーに近づくか」だけを学習すれば良いので、巨大なネットワークは要らないのです。
言い換えれば、画像認識のCNNが「ピクセルから直接、犬や猫を当てる」方式だとすると、Skalaは「犬や猫の特徴量はもう与えてある。組み合わせ方だけ学んでね」という方式です。物理学の蓄積をリスペクトしつつ、最後の難しい部分だけAIに任せる。とても賢いハイブリッド設計だと感じます。
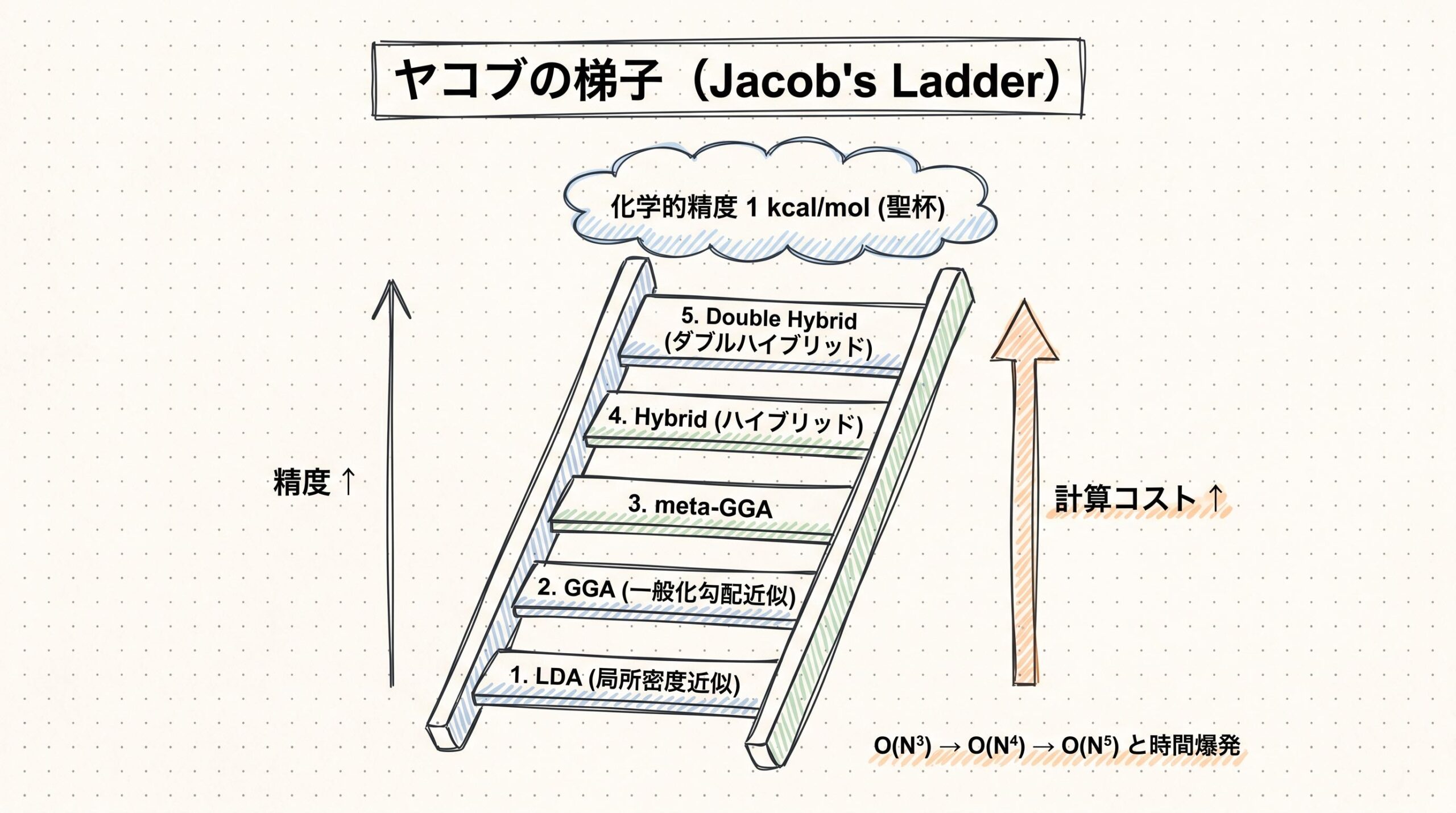
格子点同士で「おしゃべり」させる工夫
Skalaのアーキテクチャでもっとも独創的なのは、「非局所的な相関」を取り込むための仕組みです。少し噛み砕いて説明します。
DFTの計算では、分子の周りの空間を細かい「格子点」に分けて、それぞれの点で電子密度や勾配を計算します。従来のmeta-GGA汎関数は、各格子点を独立に処理していました。つまり、「ある格子点で起こっていること」と「離れた格子点で起こっていること」は無関係に扱っていたのです。
ところが、量子力学の世界では、離れた電子同士でも相互に影響しあう「非局所性」があります。これがHybrid汎関数の精度の源泉でした。Hybridは厳密交換項という重い計算でこの非局所性を取り入れていたのです。
Skalaはこれをもっと軽く実現する仕組みを発明しました。論文によれば、次のような流れになっています(論文PDFより)。
- 細かい格子点上で、meta-GGAの特徴量を計算する
- それを「原子中心の粗いグリッド」に集約する(球面調和関数でℓ=0〜3の角度成分に分解)
- 距離方向にも16次元の動径基底関数で分解する
- 原子中心グリッド上でニューラルネットが情報をやり取りする
- 処理した情報を元の細かい格子点へ逆射影する
このサイクルを3回繰り返します。原子中心グリッドが「ハブ駅」の役割を果たし、遠く離れた格子点同士でも情報交換できるようになるのです。
比喩で言えば、こんなイメージかもしれません。「会社の事務室がたくさんある中で、各部屋同士が直接話すと大変なので、まず各階のロビーに情報を集約し、ロビー同士でやりとりして、また各部屋に戻す」。情報の集約・交換・分配という構造で、計算量を抑えながら全体の事情を踏まえた処理ができるわけです。
論文の検証では、この非局所な情報交換を入れたモデルは、入れないバージョンと比べて誤差を約50%削減したことが示されています。アーキテクチャ設計が偶然うまくいったのではなく、「ここが本質だ」と狙って効いていることがわかります。
SCF微調整 物理法則をデータから自然に学ぶ
Skalaの学習プロセスにも、面白い工夫があります。二段階で訓練されているのです。
第一段階は「事前学習」。約40万件のB3LYP電子密度と、CCSD(T)/CBSという超高精度な波動関数理論データのペアで学習します。CCSD(T)は実験的にもほぼ正解とされる手法なので、「正解への写像」をニューラルネットに教える段階です。
第二段階は「SCF微調整」。SCF(Self-Consistent Field、自己無撞着場)とは、DFT計算で電子密度を繰り返し計算で収束させる手続きです。Skalaを実際のSCFループに組み込み、自分自身が出力した電子密度を使って学習を続けます。
面白いのは、このSCF微調整によって、エネルギー誤差だけでなく双極子モーメントの誤差も改善した点です。エネルギーと双極子モーメントは別の物理量なので、両方が同時に改善するということは「単なる数値合わせ」ではなく「物理的に正しい方向に学習している」証拠です。
さらに論文では、運動相関エネルギーの正値性など、教えていない物理法則をSkalaが自然に獲得していることも報告されています。「物理法則を式に埋め込まなくても、十分な高精度データがあれば物理が立ち上がってくる」。これはAlphaFoldがアミノ酸配列だけからタンパク質構造を当てる現象と似ていて、AI for Scienceの方向性を示唆しているように思います。
次のセクションでは、こうして作られたSkalaが、実際にどれほどの精度を叩き出したのかを、ベンチマーク結果の数字で確かめていきましょう。
GMTKN55ベンチマークの結果が示したもの
ここまでの説明で、Skalaの設計思想と仕組みについてはイメージできたかと思います。けれど、研究の世界では「設計が美しい」だけでは評価されません。実際にどれだけの精度が出たのか、数字で示す必要があります。このセクションでは、Skalaの実力を測ったベンチマーク結果を、具体的な数字とともに見ていきましょう。
GMTKN55とは何か 化学計算のオリンピック
DFT汎関数の性能を比べるためのベンチマークはいくつかありますが、現在もっとも信頼されているのがGMTKN55です。ドイツのGoerigk教授とGrimme教授のチームが2017年に発表した、汎関数評価の事実上の標準と言えるテストセットです。
名前の由来は「General Main-group Thermochemistry, Kinetics, and Noncovalent interactions(主族元素の熱力学、反応速度、非共有結合相互作用に関する一般的なベンチマーク)」。55種類のサブセットで構成されており、原子化エネルギー、反応エネルギー、反応障壁、分子内・分子間の弱い相互作用など、化学計算で評価される代表的な性質を網羅しています。
Goerigkチームの解説によれば、約220個の汎関数を一気に評価したことで、過去の評価研究の中でもっとも大規模な比較となりました。研究者がDFT計算で汎関数を選ぶとき、まず参照するのがこのGMTKN55の結果です。化学計算のオリンピックのような存在、と思ってもらえれば近いです。
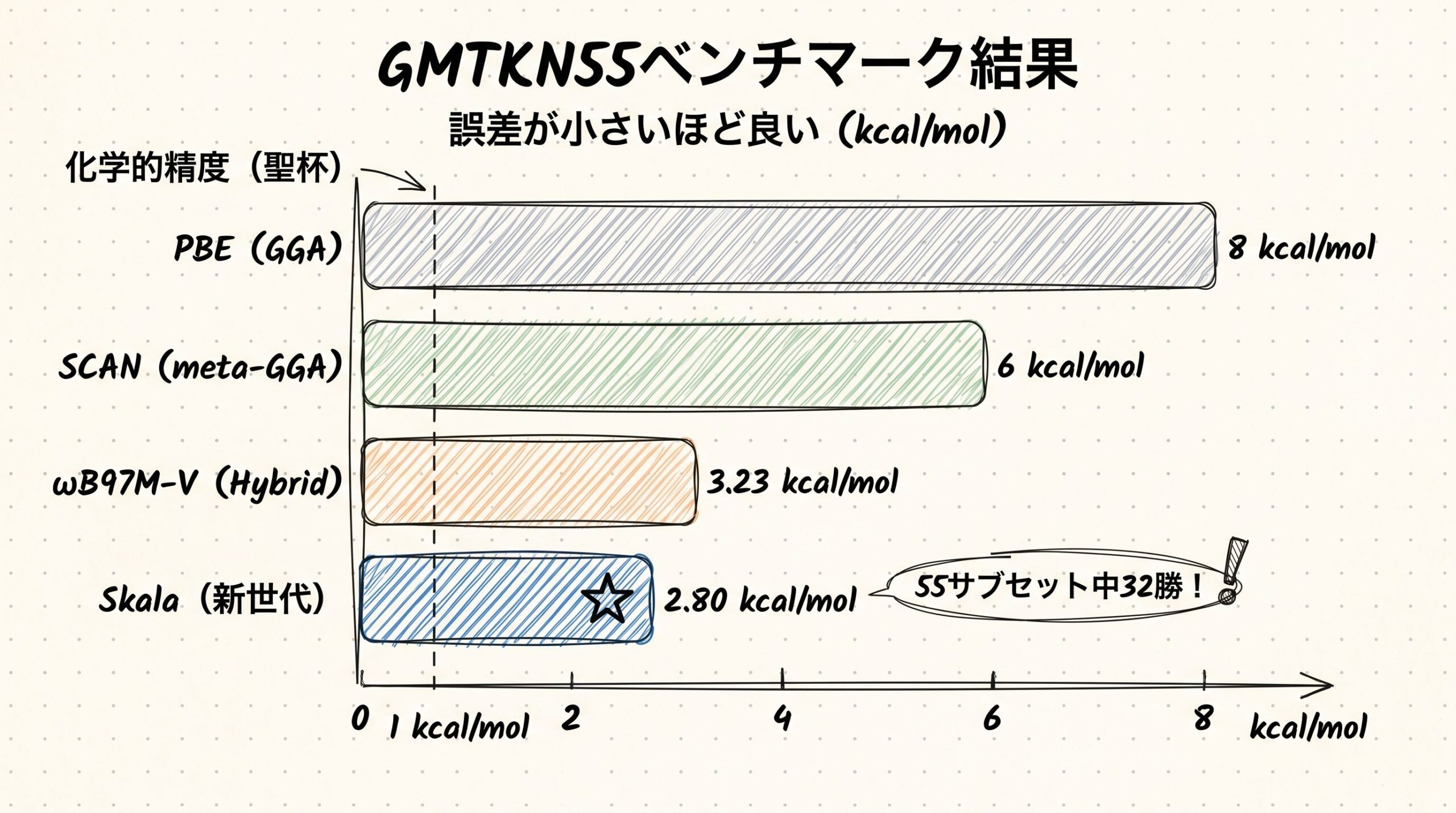
Skalaのスコアは2.80 kcal/mol
論文の本文によれば、SkalaはGMTKN55全体で平均誤差2.80 kcal/molを達成しました。さらに55個のサブセットのうち32個でトップのスコアを叩き出しています(Hugging Face要約)。
比較のために、これまでの代表的な汎関数のスコアを並べてみましょう。
| 汎関数 | 種類 | GMTKN55誤差 | 計算コスト |
|---|---|---|---|
| PBE | GGA(2段目) | 約8 kcal/mol | O(N³) |
| SCAN | meta-GGA(3段目) | 約6 kcal/mol | O(N³) |
| ωB97M-V | Hybrid(4段目) | 3.23 kcal/mol | O(N⁴)以上 |
| **Skala** | **新世代** | **2.80 kcal/mol** | **O(N³)** |
| DSD-PBEP86 | Double Hybrid(5段目) | 約2 kcal/mol | O(N⁵)以上 |
スコアだけ見ると「ωB97M-Vより0.43 kcal/mol良くなっただけ?」と感じるかもしれません。けれど、計算コストが「O(N³)のセミローカル並み」であることに注目してください。これは、原子数が増えてもHybrid汎関数のように爆発的に時間がかからないということです。
100原子の分子を計算する場面を考えてみましょう。Hybrid汎関数では数日かかる計算が、Skalaなら数時間で終わる。しかも精度は同等以上。研究者の感覚としては「数日待ち」と「数時間待ち」の差は、業務フローを根本から変えるレベルです。
「化学的精度」まであと一歩
前のセクションでもお伝えした通り、計算化学の聖杯は「化学的精度(1 kcal/mol未満)」です。Skalaの2.80 kcal/molは、まだそこには届いていません。
ですが、ここで興味深い事実があります。Microsoftチームは論文の中で、訓練データの量を増やすほどSkalaの精度が体系的に向上することを実証しました。論文要約にはこう書かれています。「modern deep learning enables systematically improvable neural exchange-correlation models as training datasets expand」。日本語に訳すと「データセットが拡大するにつれて、ニューラル交換相関モデルは体系的に改善できる」となります。
要するに、これは大規模言語モデルの世界で見られた「スケーリング則」と似た現象です。GPTシリーズが、パラメータ数とデータ量を増やすたびに性能が向上してきたように、Skalaも訓練データを増やせばさらに精度が上がる可能性が示されたのです。「化学的精度1 kcal/mol」は、もはや手の届かない目標ではなく、データ量と時間の問題に変わったのかもしれません。
Skala-1.1という改良版もすでに登場
論文の初版が2025年6月に公開されたあと、Microsoftチームは早くも改良版Skala-1.1を発表しました。Hugging Faceのモデルカードによれば、現在公開されている推奨モデルはこのSkala-1.1で、ニューヨーク大学などとの協力で生成されたデータセットで訓練されています。
論文の最新版(v6、2026年4月更新)に反映された結果を見ると、原子化エネルギーに関しては「実験レベルの精度」を達成しているとMicrosoftがAzure AI Foundryのページで報告しています。原子化エネルギーとは「分子をバラバラの原子に分解するのに必要なエネルギー」のことで、化学反応の予測の出発点となる量です。ここが実験並みに当たるのは、創薬や材料設計にとって大きな意味があります。
「速い・正確・スケールする」3拍子
改めて整理すると、Skalaが示したのは次の3つです。
- 計算速度:セミローカルDFT並みのO(N³)を維持(Hybridの10〜100倍速)
- 計算精度:GMTKN55で平均誤差2.80 kcal/mol(既存Hybridを上回り、Double Hybrid級に肉薄)
- スケーラビリティ:訓練データを増やせば精度が体系的に向上する
60年来の「精度と速さは両立しない」という常識を、データと深層学習で破った。これがSkalaの最大のインパクトだと私は感じます。
では、この技術が私たちの生活や産業にどう波及していくのでしょうか。最後のセクションで、Skalaの公開状況と、これから起きそうな未来を覗いてみましょう。
Skalaの先にある未来 創薬・電池・触媒の現場が変わる?
ここまで論文の中身を追いかけてきました。では最後に、Skalaという技術が私たちの暮らしや産業をどう変えていきそうか、少し視点を広げて考えてみたいと思います。論文単体の話を超えるのでざっくりとした話になりますが、起きつつある変化のスケッチとして読んでください。
Skalaは誰でも触れる Microsoftの「公開戦略」
まず大事な事実として、Skalaは研究目的で公開されています。
- GitHub microsoft/skala:モデル本体とコード
- Hugging Face microsoft/skala-1.1:最新の推奨モデル
- Azure AI Foundry Labs:クラウドでSkalaを試せる環境
Azure AI Foundry Labsのページには、こう書かれています。「Bringing the accuracy of DFT in line with experimental accuracy addresses a fundamental barrier to shifting the balance of molecule and material design from being driven by laboratory experiments to being driven by computational simulations」。日本語にすると「DFTの精度を実験精度に近づけることは、分子設計や材料設計の主軸を実験室から計算シミュレーションへと移行させる、根本的な障壁を取り除くことにつながる」となります。
この一文に、Microsoftの狙いが詰まっています。化学・材料の研究開発は、長らく「実験して検証する」が中心でした。シミュレーションは補助的な役割にとどまっていた。なぜなら、シミュレーションの精度が実験には及ばなかったからです。Skalaのような技術が普及すれば、その関係が逆転する可能性があります。「シミュレーションで設計し、実験は最終確認のため」という流れになっていく。
創薬の現場で起きそうなこと
新薬開発のもっとも重要なステップのひとつが、「タンパク質と薬の相互作用を正確に予測する」ことです。薬の分子が標的タンパク質にうまく結合するか、副作用を引き起こす別のタンパク質にも結合してしまわないか。これらをミリ単位、kcal単位で予測できれば、開発失敗率を劇的に下げられます。
Skalaの精度(2.80 kcal/mol、改善中)は、この用途にとってまだ十分とは言えません。けれど、化学的精度1 kcal/molに近づいていく道筋が見えたことの意味は大きいです。データを増やせば精度が上がる、という性質が確認されたからです。今後数年で、創薬向けに特化したSkala派生モデルが登場する可能性は十分にあると感じます。
電池材料探索の事例 9ヶ月で数百万候補を絞り込んだ
Microsoftはすでに、AIを使った材料探索で実績を作りつつあります。Science誌に掲載された事例では、Microsoftと太平洋北西国立研究所(PNNL)が組んで、数百万種類の電解質候補を9ヶ月で絞り込み、有望な固体電解質材料を発見しました。
この事例で使われたのは、機械学習ポテンシャル(MLポテンシャル)と呼ばれる、「DFT結果を高速に近似する」タイプのAIです。Skalaはこれとは少し役割が違っていて、DFTそのものの精度を底上げする方の技術です。両者が組み合わさると、こんな流れが現実味を帯びます。
- MLポテンシャルで、数百万から数千万の候補材料を超高速にスクリーニング
- 残った数百〜数千の有望候補を、Skalaで「実験並みの精度」で精密評価
- 最終候補を実験室で合成・検証
「広く速く探して、絞ったら精度高く検証する」という二段構えで、材料開発期間が一気に短縮されるわけです。
触媒開発でも期待大
触媒は、化学反応を加速したり選択性を高めたりする物質で、化学産業の心臓部のような存在です。Preferred Networksの技術ブログによれば、すでに同社のMatlantis製品は機械学習ポテンシャルPFPを使って触媒開発の速度を大幅に上げています。
Skalaのような新しい交換相関汎関数が普及すれば、こうした既存のAI製品の「精度の天井」も引き上げられる可能性があります。なぜなら、機械学習ポテンシャルは結局のところ「DFT計算結果」を学習して作られているからです。教師となるDFTそのものが正確になれば、生徒であるMLポテンシャルもそれに引きずられて賢くなる。教師の質が上がる、という意味で大きな波及効果が期待できます。
「DFTのImageNet時代」が始まる
論文を読んで私が一番印象に残ったのは、データ量を増やすほど精度が体系的に向上するというスケーリング則の確認でした。これは、画像認識の世界が「ImageNet以後」に変わっていったのと同じ現象が、計算化学でも起きつつあることを意味します。
2012年にAlexNetがImageNetで圧勝したあと、画像認識は「人間が特徴量を設計する時代」から「データとモデルサイズを増やす時代」へ移りました。同じ転換が、量子化学でも始まっているように見えます。論文の最終版で、Microsoftチームは「first-principles simulations to become progressively more predictive(第一原理シミュレーションが、ますます予測的になっていく)」と書いています。
計算化学が「ImageNet時代」を迎えるなら、これから5年から10年で、精度・速度・スケール、すべての面で劇的な進化が起きていく可能性が高いです。今のSkalaは、その入り口に立った最初のモデルなのかもしれません。
まとめ Skalaが教えてくれた3つのこと
長い記事になりましたが、最後に大事なポイントを整理しておきます。
- 発想の転換:手で式を作る職人芸から、データから学ぶAI職人へ。計算化学にもパラダイムシフトが起きた
- 小さくても強い:たった38万パラメータのニューラルネットが、60年積み上げてきた人類の知恵を超えた瞬間。物理的な特徴量を入力に使う賢い設計が鍵
- 誰でも触れる:GitHubとAzure AI Foundry Labsで公開され、研究者や学生が自分の手で動かせる時代がきた
論文の中身は確かに難しいです。けれど、その本質は「データと深層学習が、計算化学のあり方を根っこから変えつつある」というシンプルなメッセージにあります。中学生でも、大学生でも、プロの研究者でも、それぞれの立場でこの変化に立ち会えるのが2026年の今、という時代なのだと感じます。
もし手を動かしてみたくなったら、まずはMicrosoftのGitHubリポジトリを眺めてみるか、Hugging Faceのモデルカードを読んでみてください。クラウドで気軽に試したい方はAzure AI Foundry Labsが便利です。論文の本文はarXivで読めます。
Skalaは始まりに過ぎません。次に来るのはどんな技術なのか、私自身もとても楽しみにしています。
調査手法について
こちらの記事はグラフAIリサーチプラットフォームのSnorbeを使って作られています。Snorbeは研究開発・新規事業向けの調査テーマに応じた幅広い項目のオートリサーチや、ナレッジグラフの構築、構造化レポートの生成ができるAIリサーチツールです。
Screenshot
調査したいテーマを入力するだけで、AIが深堀りすべき観点や広げるべき調査項目をレコメンドしながら、自動でリサーチを進めます。収集した情報はナレッジグラフとして蓄積され、未調査領域(ホワイトスペース)を可視化しながら調査の網羅性を高めていけます。
また、観点マトリクスを30秒・構造化レポートを10分で自動生成する機能があり、出典付きのレポートをMarkdown/PDF形式でエクスポートできます。調査の元データも保存されるため、ファクトチェックや社内共有も容易です。
ご利用をご希望の方は、こちらよりお申し込みください。
また、グラフAIを活用した社内ナレッジ管理や、研究開発・新規事業のリサーチ支援、セルフホスト導入のご相談も受け付けています。お困りの方はお気軽にご連絡ください。
市場調査やデスクリサーチの生成AIエージェントを作っています 仲間探し中 / Founder of AI Desk Research Agent @deskrex , https://deskrex.ai


コメント